FuseMap Tutorial V: Mapping new datasets to molCCF#
In this tutorial, we will demonstrate how to map new spatial transcriptomics data to an existing FuseMap integration. This is useful when you want to analyze new samples in the context of previously integrated datasets.
FuseMap provides functionality to project new data points into the same latent space as the reference integration, allowing you to:
Compare new samples to existing integrated data
Transfer annotations and insights from the reference to new data
Analyze spatial patterns across old and new datasets together
We use our pretrained base model, and then transfer the model to slideseq.
Pre-trained base model molCCF
Slide-seq data: Langlieb et al., Nature paper
The method is similar with Tutorial IV, where you only need to change the pretrained model to molCCF.
1. Define arguments#
[10]:
import warnings
warnings.filterwarnings("ignore")
[2]:
import os
import scanpy as sc
from easydict import EasyDict as edict
from fusemap import seed_all, spatial_map
import copy
seed_all(0)
[ ]:
pretrain_model_path = "/n/netscratch/nali_lab_seas/Everyone/mingze/FuseMap_imputation/molCCF"
output_save_dir = "/n/netscratch/nali_lab_seas/Everyone/mingze/FuseMap_imputation/workspace/map_slideseq_molCCF"
args = edict(dict(pretrain_model_path=pretrain_model_path,
output_save_dir=output_save_dir,
use_llm_gene_embedding="false",
keep_celltype='',
keep_tissueregion='',
))
data_dir_list = ["/n/home11/mingzeyuan/FuseMap/data/02_imaging_sequencing_data/sequencing_test_data/slideseq_Puck34.h5ad"]
2. Data loading and pre-processing#
[4]:
X_input = []
for ind, data_dir in enumerate(data_dir_list):
print(f"Loading {data_dir}")
data = sc.read_h5ad(data_dir)
# Handle spatial coordinates
if "x" not in data.obs.columns:
if "col" in data.obs.columns and "row" in X.obs.columns:
data.obs["x"] = data.obs["col"]
data.obs["y"] = data.obs["row"]
elif "spatial" in data.obsm.keys():
data.obs["x"] = data.obsm["spatial"][:,0]
data.obs["y"] = data.obsm["spatial"][:,1]
elif 'Raw_Slideseq_X' in data.obs.columns:
data.obs['x'] = data.obs['Raw_Slideseq_X']
data.obs['y'] = data.obs['Raw_Slideseq_Y']
else:
raise ValueError(f"Please provide spatial coordinates in the obs['x'] and obs['y'] columns for {data_dir}")
# Add metadata
data.obs['name'] = f'section{ind}'
data.obs['file_name'] = os.path.basename(data_dir)
print(f"Loaded {data.shape[0]} cells with {data.shape[1]} genes from {data.obs['file_name'].iloc[0]}")
X_input.append(data)
# Set parameters for integration
kneighbor = ["delaunay"] * len(X_input)
input_identity = ["ST"] * len(X_input)
print(f"Loaded {len(X_input)} datasets")
Loading /n/home11/mingzeyuan/FuseMap/data/02_imaging_sequencing_data/sequencing_test_data/slideseq_Puck34.h5ad
Loaded 72542 cells with 20543 genes from slideseq_Puck34.h5ad
Loaded 1 datasets
3. Mapping fusemap to new datasets#
[5]:
for i in range(len(X_input)):
args_i=copy.copy(args)
args_i.output_save_dir = os.path.join(args.output_save_dir, X_input[i].obs['file_name'].unique()[0])
spatial_map([X_input[i]], args_i, [kneighbor[i]], [input_identity[i]])
93%|█████████▎| 14/15 [20:14<01:44, 104.65s/it]
Epoch 00004: reducing learning rate of group 0 to 5.0000e-04.
Epoch 00004: reducing learning rate of group 0 to 5.0000e-04.
100%|██████████| 15/15 [22:07<00:00, 88.50s/it]
100%|██████████| 1134/1134 [00:18<00:00, 60.06it/s]
4. Transferring annotations#
[6]:
output_dir = os.path.join(args.output_save_dir, X_input[0].obs['file_name'].unique()[0])
[7]:
from fusemap.utils import NNTransferTrain, NNTransfer, NNTransferPredictWithUncertainty
import torch
from sklearn import preprocessing
from torch.utils.data import DataLoader, TensorDataset
from torch.utils.data import random_split
from torch import optim, nn
import sklearn
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
import pandas as pd
import pickle
[14]:
def transfer_annotation(molccf_path, save_dir):
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
### transfer cell type
# Load the .pkl file
ad_embed = sc.read_h5ad(os.path.join(save_dir, 'ad_celltype_embedding.h5ad'))
if 'fusemap_celltype' not in ad_embed.obs.columns:
with open(os.path.join(molccf_path, 'transfer', 'le_gt_cell_type_main_STARmap.pkl'), 'rb') as file:
le_gt_cell_type_main_STARmap = pickle.load(file)
NNmodel = NNTransfer(input_dim=64,output_dim=len(le_gt_cell_type_main_STARmap))
NNmodel.load_state_dict(torch.load(molccf_path+"/transfer/NNtransfer_cell_type_main_STARmap.pt"))
dataset = TensorDataset(torch.Tensor(ad_embed.X))
dataloader = DataLoader(dataset, batch_size=256, shuffle=False)
NNmodel.to(device)
NNmodel.eval()
all_predictions = []
with torch.no_grad():
for inputs in dataloader:
inputs = inputs[0].to(device)
outputs = NNmodel(inputs)
_, predicted = torch.max(outputs, 1)
all_predictions.extend(predicted.detach().cpu().numpy())
ad_embed.obs['fusemap_celltype']=[le_gt_cell_type_main_STARmap[i] for i in all_predictions]
ad_embed.write_h5ad(save_dir + "/ad_celltype_embedding.h5ad")
### transfer tissue niche
# Load the .pkl file
ad_embed = sc.read_h5ad(save_dir + "/ad_tissueregion_embedding.h5ad")
if 'fusemap_tissueregion' not in ad_embed.obs.columns:
with open(os.path.join(molccf_path, 'transfer', 'le_gt_tissue_region_main_STARmap.pkl'), 'rb') as file:
le_gt_tissue_region_main_STARmap = pickle.load(file)
NNmodel = NNTransfer(input_dim=64,output_dim=len(le_gt_tissue_region_main_STARmap))
NNmodel.load_state_dict(torch.load(molccf_path+"/transfer/NNtransfer_tissue_region_main_STARmap.pt"))
dataset = TensorDataset(torch.Tensor(ad_embed.X))
dataloader = DataLoader(dataset, batch_size=256, shuffle=False)
NNmodel.to(device)
NNmodel.eval()
all_predictions = []
with torch.no_grad():
for inputs in dataloader:
inputs = inputs[0].to(device)
outputs = NNmodel(inputs)
_, predicted = torch.max(outputs, 1)
all_predictions.extend(predicted.detach().cpu().numpy())
ad_embed.obs['fusemap_tissueregion']=[le_gt_tissue_region_main_STARmap[i] for i in all_predictions]
ad_embed.write_h5ad(save_dir + "/ad_tissueregion_embedding.h5ad")
[15]:
transfer_annotation(pretrain_model_path, output_dir)
5. Plot transferred cell types and regions#
[16]:
ad_fusemap_transfer = sc.read_h5ad(os.path.join(output_dir, 'ad_celltype_embedding.h5ad'))
cell_types = sorted(ad_fusemap_transfer.obs['fusemap_celltype'].unique())
# Generate unique colors using a continuous colormap
num_colors = len(cell_types)
cmap = plt.get_cmap('gist_rainbow', num_colors) # 'gist_rainbow' ensures distinct colors
colors = [cmap(i / num_colors) for i in range(num_colors)]
# Create a dictionary mapping tissue types to colors
colormap = dict(zip(cell_types, colors))
plt.rcParams['figure.figsize'] = (8, 8)
plt.rcParams['figure.dpi'] = 300
# Get coordinates for sample
x = pd.to_numeric(ad_fusemap_transfer.obs['x'], errors='coerce')
y = pd.to_numeric(ad_fusemap_transfer.obs['y'], errors='coerce')
# Calculate centroid
centroid = (np.mean(x), np.mean(y))
# Center points
x_centered = x - centroid[0]
y_centered = y - centroid[1]
# Calculate scale factor to normalize points
scale = np.sqrt(np.mean(x_centered**2 + y_centered**2))
# Scale points to normalize spread
x_normalized = x_centered / scale
y_normalized = y_centered / scale
# Stack coordinates
coords = np.column_stack((x_normalized, y_normalized))
# Create a figure with subplots for each tissue type
n_tissues = len(cell_types)
n_cols = 4
n_rows = (n_tissues + n_cols - 1) // n_cols
fig, axes = plt.subplots(n_rows, n_cols, figsize=(20, 5*n_rows), dpi=300)
axes = axes.flatten()
# Plot each tissue type separately
for idx, tissue in enumerate(cell_types):
ax = axes[idx]
mask = ad_fusemap_transfer.obs['fusemap_celltype'] == tissue
# Plot other cells with transparency
ax.scatter(coords[~mask, 0], coords[~mask, 1], s=0.3,
c='gray', alpha=0.1)
# Plot cells of current tissue type
ax.scatter(coords[mask, 0], coords[mask, 1], s=0.3,
c=[colormap[tissue]], label=tissue)
ax.set_title(tissue)
ax.axis('off')
# Remove any empty subplots
for idx in range(n_tissues, len(axes)):
fig.delaxes(axes[idx])
plt.tight_layout()
plt.show()
[17]:
ad_fusemap_transfer = sc.read_h5ad(os.path.join(output_dir, 'ad_tissueregion_embedding.h5ad'))
tissue_regions = sorted(ad_fusemap_transfer.obs['fusemap_tissueregion'].unique())
# Generate unique colors using a continuous colormap
num_colors = len(tissue_regions)
cmap = plt.get_cmap('gist_rainbow', num_colors) # 'gist_rainbow' ensures distinct colors
colors = [cmap(i / num_colors) for i in range(num_colors)]
# Create a dictionary mapping tissue types to colors
colormap = dict(zip(tissue_regions, colors))
plt.rcParams['figure.figsize'] = (8, 8)
plt.rcParams['figure.dpi'] = 300
# Get coordinates for sample
x = pd.to_numeric(ad_fusemap_transfer.obs['x'], errors='coerce')
y = pd.to_numeric(ad_fusemap_transfer.obs['y'], errors='coerce')
# Calculate centroid
centroid = (np.mean(x), np.mean(y))
# Center points
x_centered = x - centroid[0]
y_centered = y - centroid[1]
# Calculate scale factor to normalize points
scale = np.sqrt(np.mean(x_centered**2 + y_centered**2))
# Scale points to normalize spread
x_normalized = x_centered / scale
y_normalized = y_centered / scale
# Stack coordinates
coords = np.column_stack((x_normalized, y_normalized))
# Create a figure with subplots for each tissue region
n_regions = len(tissue_regions)
n_cols = 4
n_rows = (n_regions + n_cols - 1) // n_cols
fig, axes = plt.subplots(n_rows, n_cols, figsize=(20, 5*n_rows), dpi=300)
axes = axes.flatten()
# Plot each tissue region separately
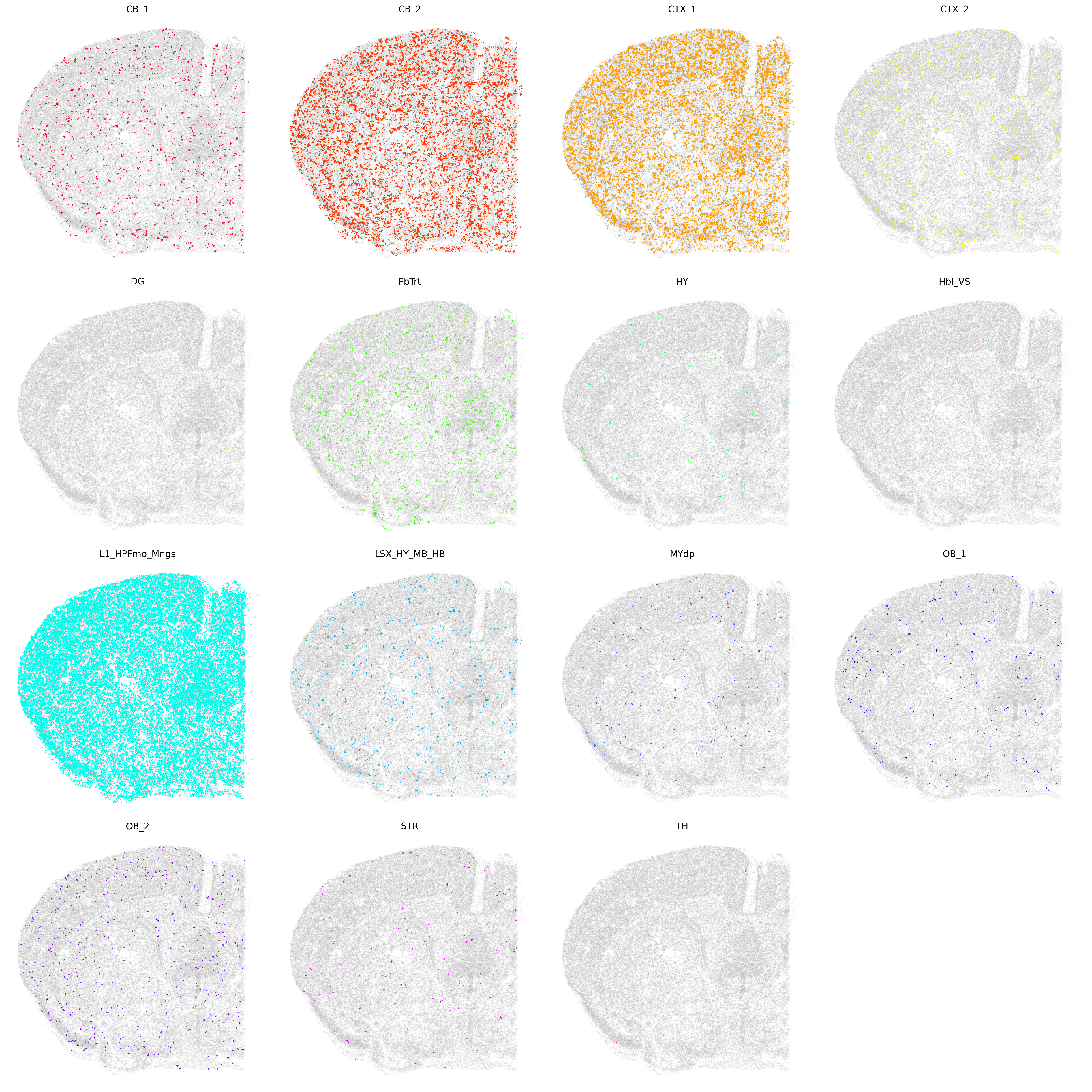
for idx, region in enumerate(tissue_regions):
ax = axes[idx]
mask = ad_fusemap_transfer.obs['fusemap_tissueregion'] == region
# Plot other cells with transparency
ax.scatter(coords[~mask, 0], coords[~mask, 1], s=0.3,
c='gray', alpha=0.1)
# Plot cells of current tissue region
ax.scatter(coords[mask, 0], coords[mask, 1], s=0.3,
c=[colormap[region]], label=region)
ax.set_title(region)
ax.axis('off')
# Remove any empty subplots
for idx in range(n_regions, len(axes)):
fig.delaxes(axes[idx])
plt.tight_layout()
plt.show()