Fusemap tutorial VI: Exploring cell-to-cell interactions#
[1]:
import warnings
warnings.filterwarnings("ignore")
[2]:
import os
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as sc
import statsmodels.stats.multitest
[ ]:
# At the beginning of the notebook, after imports
SOURCE_DATA_BASE = '/n/netscratch/nali_lab_seas/Everyone/mingze/FuseMap_imputation/tutorial6_source_data'
# Define common subdirectories
def get_paths(focus_key):
paths = {
'ct_labels': f'{SOURCE_DATA_BASE}/df_ct_labels_{focus_key}.csv',
'cells_by_regions': f'{SOURCE_DATA_BASE}/cells_by_regions_{focus_key}',
'outputs_30um': f'{SOURCE_DATA_BASE}/outputs_30um_{focus_key}',
'color': f'{SOURCE_DATA_BASE}/color/starmap_sub.csv'
}
return paths
1. Get cells by regions#
[4]:
def get_confusion_count_df(df, col1, col2):
'''Get the confusion matrix between two categorical columns in a dataframe.'''
assert((col1 != 'count') and (col2 != 'count'))
count_df = df[[col1, col2]].copy()
count_df['count'] = 1
conf_df = pd.pivot_table(count_df, index=[col1], columns=[col2],
values='count', aggfunc=np.sum).fillna(0)
return conf_df
def get_expected_count_df(conf_df):
mtx = conf_df.values
total_count = np.sum(mtx)
row_fractions = np.sum(mtx, axis=1) / total_count
col_fractions = np.sum(mtx, axis=0) / total_count
expect_fractions = row_fractions[:, np.newaxis] * col_fractions[np.newaxis, :]
expected_count_df = pd.DataFrame(data=expect_fractions * total_count,
index=conf_df.index, columns=conf_df.columns)
return expected_count_df
[5]:
for focus_key in ['Atlas1','Atlas2','Atlas3']:
df_ct_labels=pd.read_csv(f'{SOURCE_DATA_BASE}/df_ct_labels_{focus_key}.csv',index_col=0)
output_path = f'{SOURCE_DATA_BASE}/cells_by_regions_{focus_key}'
os.makedirs(output_path, exist_ok=True)
# Get the confusion matrix data frame
conf_df = get_confusion_count_df(df_ct_labels, 'transfer_gt_cell_type_sub_STARmap', 'transfer_gt_tissue_region_main_STARmap')
# Calculate the enrichment matrix data frame
expected_count_df = get_expected_count_df(conf_df)
region_enrichment_df = conf_df / expected_count_df.values
major_brain_regions = list(df_ct_labels['transfer_gt_tissue_region_main_STARmap'].unique())
for r in major_brain_regions:
print(r)
region_df_ct_labels = df_ct_labels[df_ct_labels['transfer_gt_tissue_region_main_STARmap'] == r]
subclasses, counts = np.unique(region_df_ct_labels['transfer_gt_cell_type_sub_STARmap'], return_counts=True)
selected_subclasses = []
for i in np.argsort(-counts):
selected = False
neuron_cattegory_label=df_ct_labels.loc[df_ct_labels['transfer_gt_cell_type_sub_STARmap']==subclasses[i],'neuron_category'].unique()[0]
# For non-neuronal, non-astrocytes
if neuron_cattegory_label=='non' and (not subclasses[i].startswith('AC')):
if counts[i] > 50:
selected = True
selected_subclasses.append(subclasses[i])
# For astrocytes
elif subclasses[i].startswith('AC'):
if region_enrichment_df.loc[subclasses[i], r] > 1:
selected = True
selected_subclasses.append(subclasses[i])
# For neurons
else:
threshold = 6
if region_enrichment_df.loc[subclasses[i], r] > threshold:
selected = True
selected_subclasses.append(subclasses[i])
region_df_ct_labels = region_df_ct_labels[region_df_ct_labels['transfer_gt_cell_type_sub_STARmap'].isin(
selected_subclasses)].copy()
region_df_ct_labels.to_csv(os.path.join(output_path, f'{r}.csv'))
L1_HPFmo_Mngs
CTX_1
CTX_2
FbTrt
STR
LSX_HY_MB_HB
OB_1
Hbl_VS
HPF_CA
DG
ENTm
CB_2
CB_1
TH
OB_2
MYdp
HY
LSX_HY_MB_HB
FbTrt
CB_1
CB_2
MYdp
Hbl_VS
L1_HPFmo_Mngs
TH
CTX_1
CTX_2
ENTm
DG
HPF_CA
HY
OB_1
STR
OB_2
OB_2
OB_1
L1_HPFmo_Mngs
CTX_2
FbTrt
CTX_1
LSX_HY_MB_HB
STR
Hbl_VS
HPF_CA
TH
DG
HY
ENTm
CB_2
CB_1
MYdp
2. Randomize and count cell-cell contacts within 30µm#
[6]:
import os
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import anndata
import scanpy as sc
sc.settings.n_jobs = 56
sc.settings.set_figure_params(dpi=180, dpi_save=300, frameon=False, figsize=(4, 4), fontsize=8, facecolor='white')
from tqdm import tqdm
import sys
sys.path.append('.')
from fusemap.permutation import generate_cell_type_contact_count_matrices
Count cell-cell contacts at the subclass level
[7]:
major_brain_regions = ['LSX_HY_MB_HB',
'FbTrt',
'CB_1',
'CB_2',
'MYdp',
'Hbl_VS',
'L1_HPFmo_Mngs',
'TH',
'CTX_1',
'CTX_2',
'ENTm',
'DG',
'HPF_CA',
'HY',
'OB_1',
'STR',
'OB_2']
[ ]:
# Make the output path
for focus_key in ['Atlas1','Atlas2','Atlas3']:
output_path = f'{SOURCE_DATA_BASE}/outputs_30um_{focus_key}'
os.makedirs(output_path, exist_ok=True)
r_radius=1.997*2
r_permute_radius=13.315
for region in major_brain_regions:
print(region)
# Read the data
df_ct_labels = pd.read_csv(os.path.join(f'{SOURCE_DATA_BASE}/cells_by_regions_{focus_key}', f'{region}.csv'), index_col=0)
slice_ids = np.unique(df_ct_labels['ap_order'])
cell_type_col = 'transfer_gt_cell_type_sub_STARmap'
cell_types = np.unique(df_ct_labels[cell_type_col])
N_cell_types = len(cell_types)
N_permutations = 1000
# Count and save the contacts without permutation
merged_contact_counts = np.zeros((N_cell_types, N_cell_types), dtype=int)
for slice_id in tqdm(slice_ids):
df_slice = df_ct_labels[df_ct_labels['ap_order'] == slice_id]
cell_type_contact_counts = generate_cell_type_contact_count_matrices(df_slice, cell_type_col,
['use_x', 'use_y'], cell_types,
permutation_method='no_permutation', contact_radius=r_radius)
merged_contact_counts = merged_contact_counts + cell_type_contact_counts
output_file = os.path.join(output_path, f'{region}_no_permutation.npy')
np.save(output_file, merged_contact_counts)
from multiprocessing import Pool
def permute_and_count_contacts_for_slices(df_slice_list):
merged_contact_counts = np.zeros((N_permutations, N_cell_types, N_cell_types), dtype=int)
for df_slice in df_slice_list:
for i in tqdm(range(N_permutations)):
df_slice_rand = df_slice.copy()
r_permute = r_permute_radius
r = r_permute * np.sqrt(np.random.uniform(size=df_slice_rand.shape[0]))
theta = np.random.uniform(size=df_slice_rand.shape[0]) * 2 * np.pi
df_slice_rand['use_x'] += r * np.sin(theta)
df_slice_rand['use_y'] += r * np.cos(theta)
cell_type_contact_counts = generate_cell_type_contact_count_matrices(df_slice_rand, cell_type_col,
['use_x', 'use_y'], cell_types,
permutation_method='no_permutation', contact_radius=r_radius)
merged_contact_counts[i] = merged_contact_counts[i] + cell_type_contact_counts
return merged_contact_counts
# Get the dataframe for each slice
all_df_slice_list = [df_ct_labels[df_ct_labels['ap_order'] == slice_id] for slice_id in slice_ids]
# Split the slices into groups
N_groups = 16
group_size = int(np.ceil(len(slice_ids) / N_groups))
grouped_slice_dfs = []
for i in range(N_groups):
slice_id_start = i * group_size
slice_id_stop = (i + 1) * group_size
grouped_slice_dfs.append(all_df_slice_list[slice_id_start:slice_id_stop])
# Permute and count the contacts in parallel
print('start')
with Pool(N_groups) as p:
contact_analysis_results = p.map(permute_and_count_contacts_for_slices, grouped_slice_dfs)
merged_contact_counts = np.sum(contact_analysis_results, axis=0)
means = np.mean(merged_contact_counts, axis=0)
stds = np.std(merged_contact_counts, axis=0)
np.save(os.path.join(output_path, f'{region}_local_permutation_count_tensor.npy'),
merged_contact_counts)
output_file_mean = os.path.join(output_path, f'{region}_local_permutation_mean.npy')
np.save(output_file_mean, means)
output_file_std = os.path.join(output_path, f'{region}_local_permutation_std.npy')
np.save(output_file_std, stds)
3. Get significant contacts within 30µm#
[9]:
def count_zero_pairs(contact_mtx):
n_0 = 0
for i in range(contact_mtx.shape[0]):
for j in range(i, contact_mtx.shape[0]):
if contact_mtx[i, j] == 0:
n_0 += 1
return n_0
def adjust_p_value_matrix_by_BH(p_val_mtx):
'''Adjust the p-values in a matrix by the Benjamini/Hochberg method.
The matrix should be symmetric.
'''
p_val_sequential = []
N = p_val_mtx.shape[0]
for i in range(N):
for j in range(i, N):
p_val_sequential.append(p_val_mtx[i, j])
p_val_sequential_bh = statsmodels.stats.multitest.multipletests(p_val_sequential, method='fdr_bh')[1]
adjusted_p_val_mtx = np.zeros((N, N))
counter = 0
for i in range(N):
for j in range(i, N):
adjusted_p_val_mtx[i, j] = p_val_sequential_bh[counter]
adjusted_p_val_mtx[j, i] = p_val_sequential_bh[counter]
counter += 1
return adjusted_p_val_mtx
def get_data_frame_from_metrices(cell_types, mtx_dict):
N = len(cell_types)
serials_dict = {'cell_type1':[], 'cell_type2':[]}
for k in mtx_dict.keys():
serials_dict[k] = []
for i in range(N):
for j in range(i, N):
serials_dict['cell_type1'].append(cell_types[i])
serials_dict['cell_type2'].append(cell_types[j])
for k in mtx_dict.keys():
serials_dict[k].append(mtx_dict[k][i, j])
return pd.DataFrame(serials_dict)
def sort_cell_type_contact_p_values(p_val_mtx, cell_types):
'''Return a list of (cell_type1, cell_type2, p_value) sorted by p_values.'''
p_val_list = []
N = p_val_mtx.shape[0]
for i in range(N):
for j in range(i, N):
p_val_list.append((cell_types[i], cell_types[j], p_val_mtx[i, j]))
return sorted(p_val_list, key=lambda x:x[2])
[10]:
import scipy.cluster
# from scattermap import scattermap
def get_optimal_order_of_mtx(X):
Z = scipy.cluster.hierarchy.ward(X)
return scipy.cluster.hierarchy.leaves_list(
scipy.cluster.hierarchy.optimal_leaf_ordering(Z, X))
def get_ordered_tick_labels(tick_labels):
tick_labels_with_class = [s.split(' ')[-1] + ' ' + s for s in tick_labels]
return np.argsort(tick_labels_with_class)
def filter_pval_mtx(pval_mtx, tick_labels, allowed_pairs):
pval_mtx_filtered = pval_mtx.copy()
for i in range(pval_mtx.shape[0]):
ct1 = tick_labels[i]
for j in range(pval_mtx.shape[1]):
ct2 = tick_labels[j]
if ((ct1, ct2) in allowed_pairs) or ((ct2, ct1) in allowed_pairs):
continue
else:
pval_mtx_filtered[i, j] = 1
return pval_mtx_filtered
[11]:
def make_dotplot(pval_mtx, fold_change_mtx, tick_labels, title='', allowed_pairs=None):
#optimal_order = get_optimal_order_of_mtx(pval_mtx)
optimal_order = get_ordered_tick_labels(tick_labels)
pval_mtx = pval_mtx[optimal_order][:, optimal_order]
fold_change_mtx = fold_change_mtx[optimal_order][:, optimal_order]
tick_labels = tick_labels[optimal_order]
if None is not allowed_pairs:
pval_mtx = filter_pval_mtx(pval_mtx, tick_labels, allowed_pairs)
pval_mtx[pval_mtx>0.05]=1
mlog_pvals = - np.log10(np.maximum(pval_mtx, 1e-10))
fold_change_mtx[mlog_pvals==0]=0
fold_change_mtx=np.log10(fold_change_mtx+1)*100
fig_len = len(tick_labels) * 0.1
# fig = plt.figure(figsize=(fig_len, fig_len), dpi=300)
fig,ax = scattermap(mlog_pvals, marker_size= fold_change_mtx,
square=True,
cmap="Reds",
linewidths=0.2 * (pval_mtx < 0.05).reshape(-1),
linecolor='black', xticklabels=tick_labels, yticklabels=tick_labels,
vmin=0, vmax=np.max(mlog_pvals),
cbar_kws={'shrink':0.5, 'anchor':(0, 0.7)})
plt.tight_layout()
fig.savefig(f'figures_{focus_key}/{title}.png',dpi=300)#, transparent=True)
import numpy as np
import matplotlib.pyplot as plt
from matplotlib.colors import Normalize
def scattermap(data_matrix, marker_size, square=True, cmap='coolwarm',
linewidths=0, linecolor='black', xticklabels=None, yticklabels=None,
vmin=None, vmax=None, cbar_kws=None):
if vmin is None:
vmin = data_matrix.min()
if vmax is None:
vmax = data_matrix.max()
norm = Normalize(vmin=vmin, vmax=vmax)
fig, ax = plt.subplots()
cmap = plt.get_cmap(cmap)
# Plot each data point individually
n, m = data_matrix.shape
for i in range(n):
for j in range(m):
color = cmap(norm(data_matrix[i, j]))
size = marker_size[i, j] if marker_size.shape == data_matrix.shape else marker_size
ax.scatter(j, i, color=color, s=size)
# Customizations
ax.set_xticks(np.arange(m))
ax.set_yticks(np.arange(n))
ax.set_xticklabels(xticklabels if xticklabels is not None else np.arange(m), rotation=90)
ax.set_yticklabels(yticklabels if yticklabels is not None else np.arange(n))
ax.invert_yaxis()
# Gridlines based on the data positions
ax.set_xticks(np.arange(m+1)-.5, minor=True)
ax.set_yticks(np.arange(n+1)-.5, minor=True)
# ax.grid(which="minor", color="w", linestyle='-', linewidth=2)
ax.tick_params(which="minor", size=0)
# Colorbar
if cbar_kws is None:
cbar_kws = {}
sm = plt.cm.ScalarMappable(cmap=cmap, norm=norm)
sm.set_array([])
cbar = plt.colorbar(sm, ax=ax, **cbar_kws)
cbar.set_label('log p value', rotation=270, labelpad=15)
if square:
plt.axis('equal')
# plt.show()
return fig,ax
[12]:
for focus_key in ['Atlas1','Atlas1','Atlas1']:
permutation_path = f'{SOURCE_DATA_BASE}/outputs_30um_{focus_key}'
os.makedirs(permutation_path+'/result/', exist_ok=True)
result_dfs = []
for region in major_brain_regions:
if os.path.exists(os.path.join(permutation_path, f'{region}_local_permutation_mean.npy')):
print(region)
# Load the cell type labels
df_ct_labels = pd.read_csv(os.path.join(f'{SOURCE_DATA_BASE}/cells_by_regions_{focus_key}', f'{region}.csv'), index_col=0)
subclass_types = np.unique(df_ct_labels['transfer_gt_cell_type_sub_STARmap'])
cell_contact_counts = np.load(os.path.join(permutation_path, f'{region}_no_permutation.npy'))
local_null_means = np.load(os.path.join(permutation_path, f'{region}_local_permutation_mean.npy'))
local_null_stds = np.load(os.path.join(permutation_path, f'{region}_local_permutation_std.npy'))
# Require all stds to be larger or equal to the minimal observable std value
local_null_stds = np.maximum(local_null_stds, np.sqrt(1 / 1000))
local_z_scores = (cell_contact_counts - local_null_means) / local_null_stds
local_p_values = scipy.stats.norm.sf(local_z_scores)
adjusted_local_p_values = adjust_p_value_matrix_by_BH(local_p_values)
fold_changes = cell_contact_counts / (local_null_means + 1e-4)
# Gather all results into a data frame
contact_result_df = get_data_frame_from_metrices(subclass_types,
{'pval-adjusted': adjusted_local_p_values,
'pval': local_p_values,
'z_score': local_z_scores,
'contact_count': cell_contact_counts,
'permutation_mean': local_null_means,
'permutatmerion_std': local_null_stds
}).sort_values('z_score', ascending=False)
# Filter out pairs that don't contact
contact_result_df = contact_result_df[contact_result_df['pval-adjusted'] < 0.05]
contact_result_df = contact_result_df[contact_result_df['contact_count'] > 50]
contact_result_df.to_csv(os.path.join(permutation_path+'/result/', f'{region}_close_contacts.csv'))
contact_result_df['region']=region
result_dfs.append(contact_result_df)
else:
print(region,'norun')
combined_results = pd.concat(result_dfs)
combined_results.to_csv(os.path.join(permutation_path+'/result/', 'all_close_contacts.csv'))
LSX_HY_MB_HB
FbTrt
CB_1
CB_2
MYdp
Hbl_VS
L1_HPFmo_Mngs
TH
CTX_1
CTX_2
ENTm
DG
HPF_CA
HY
OB_1
STR
OB_2
LSX_HY_MB_HB
FbTrt
CB_1
CB_2
MYdp
Hbl_VS
L1_HPFmo_Mngs
TH
CTX_1
CTX_2
ENTm
DG
HPF_CA
HY
OB_1
STR
OB_2
LSX_HY_MB_HB
FbTrt
CB_1
CB_2
MYdp
Hbl_VS
L1_HPFmo_Mngs
TH
CTX_1
CTX_2
ENTm
DG
HPF_CA
HY
OB_1
STR
OB_2
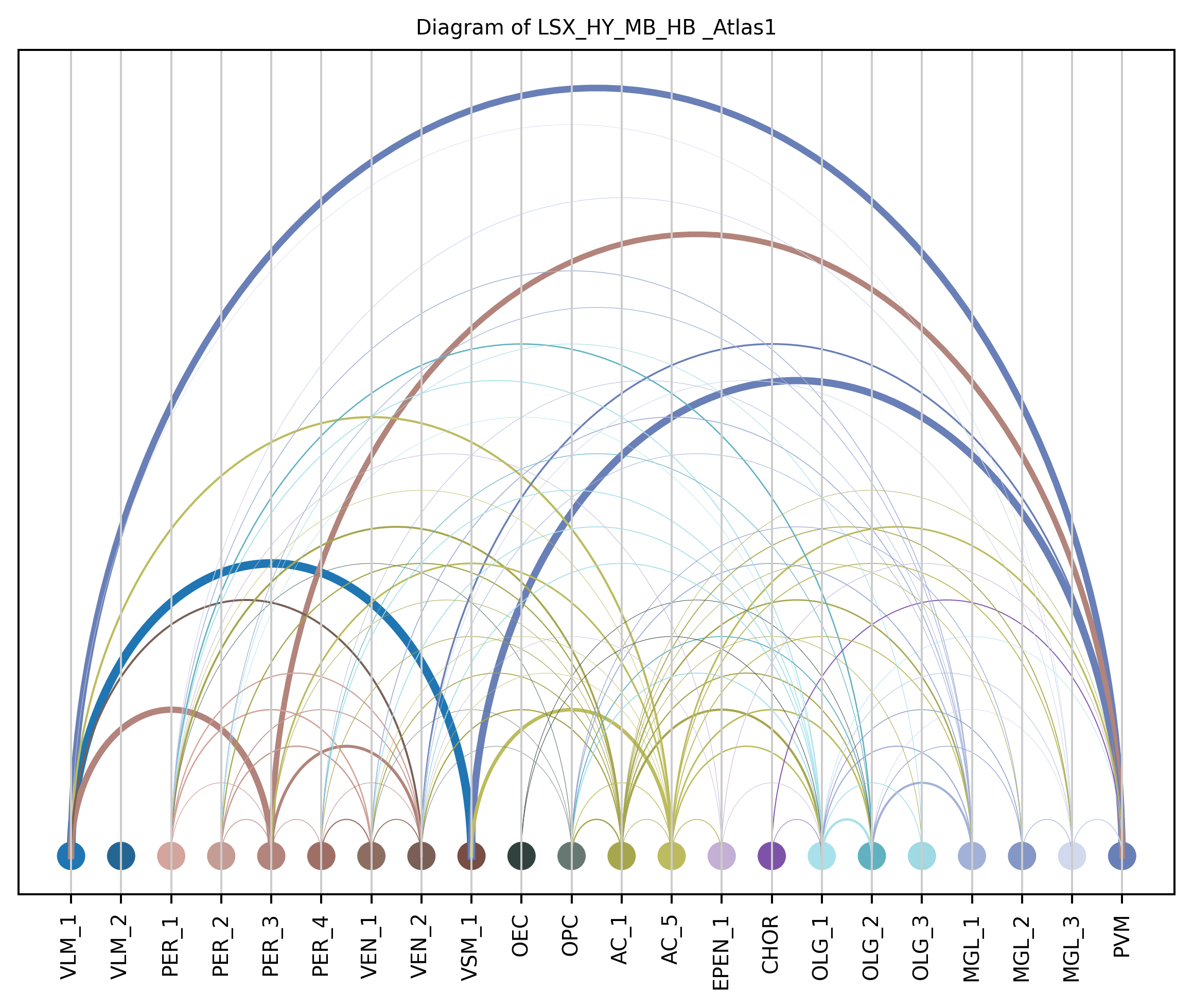
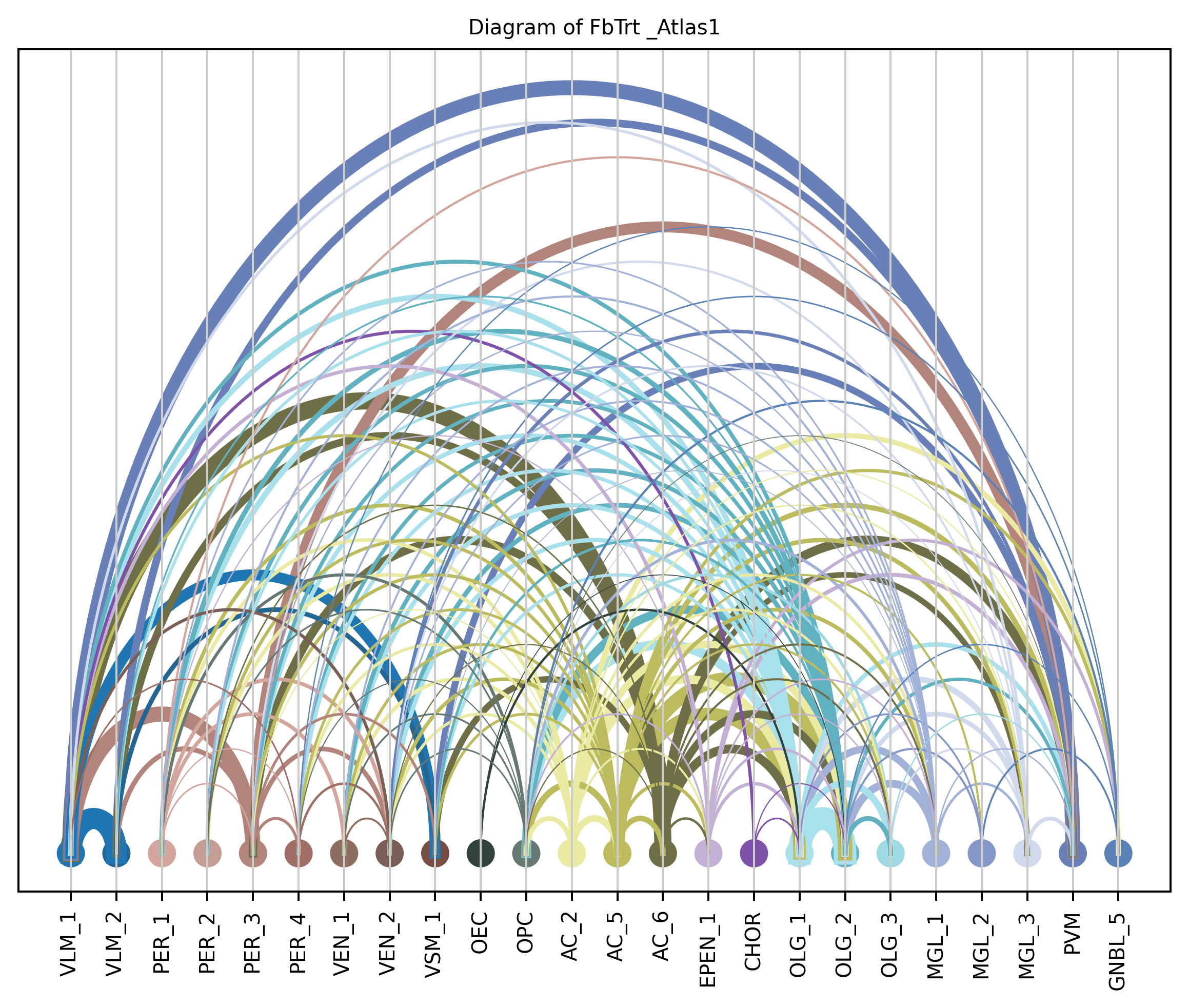
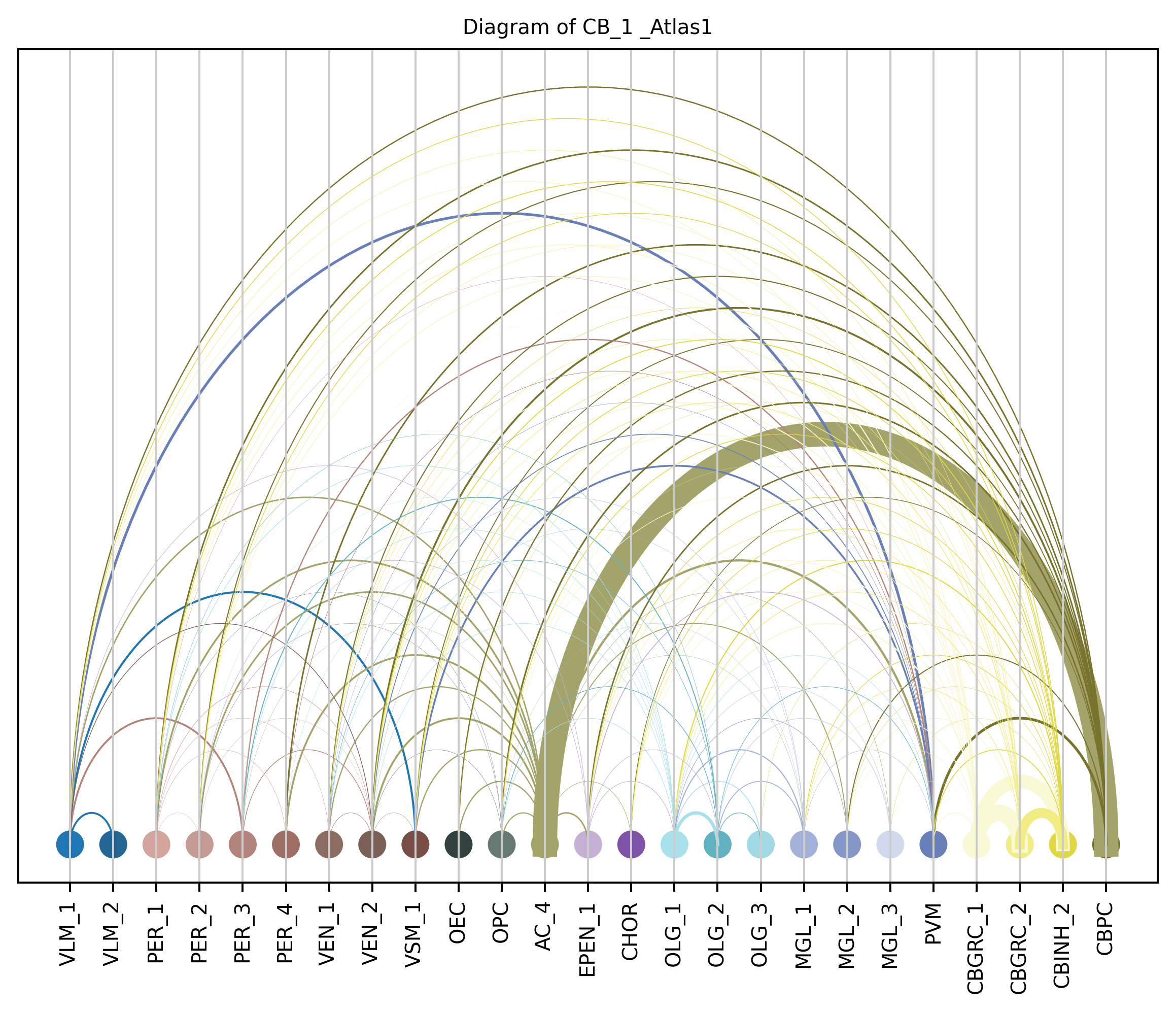
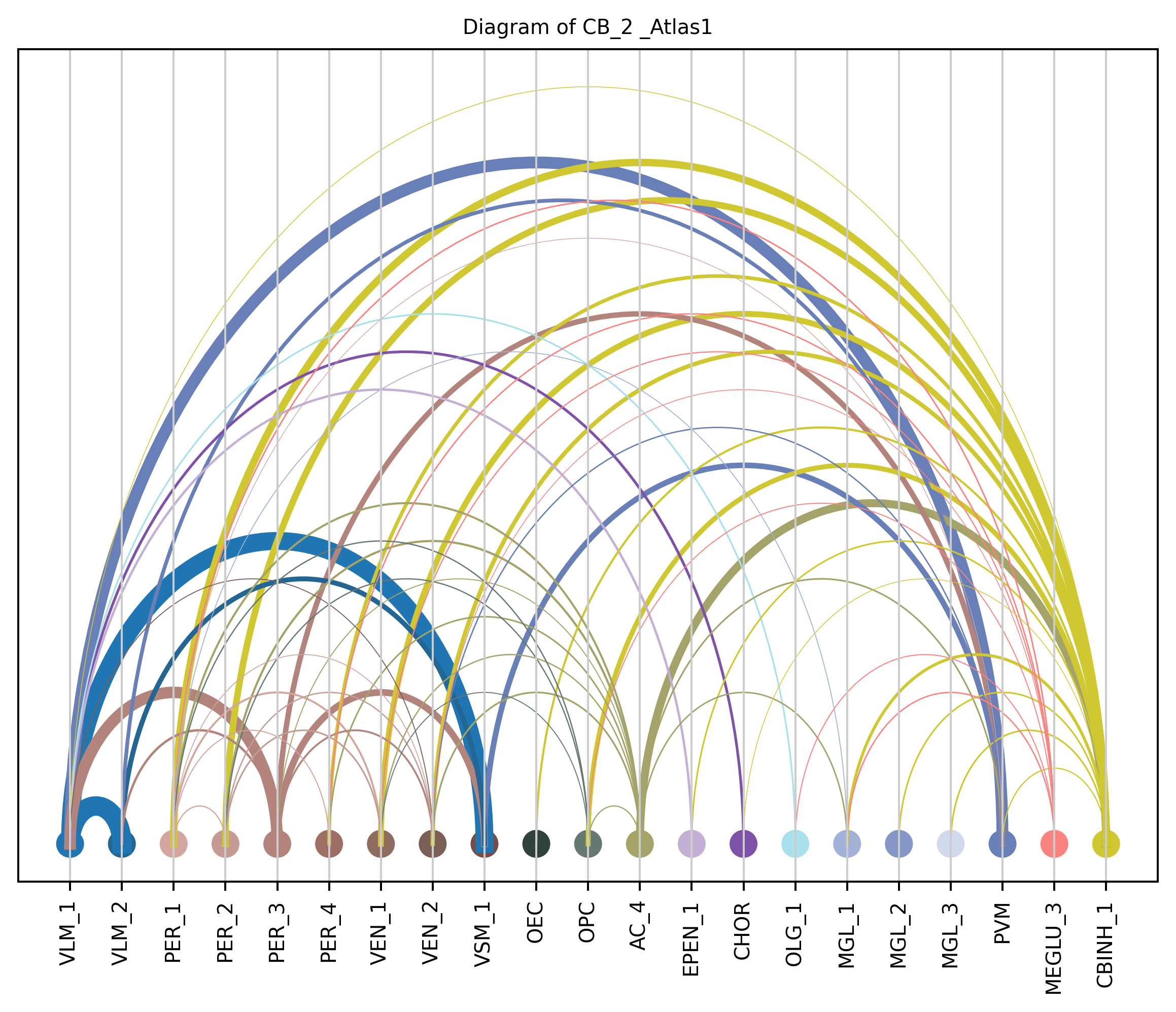
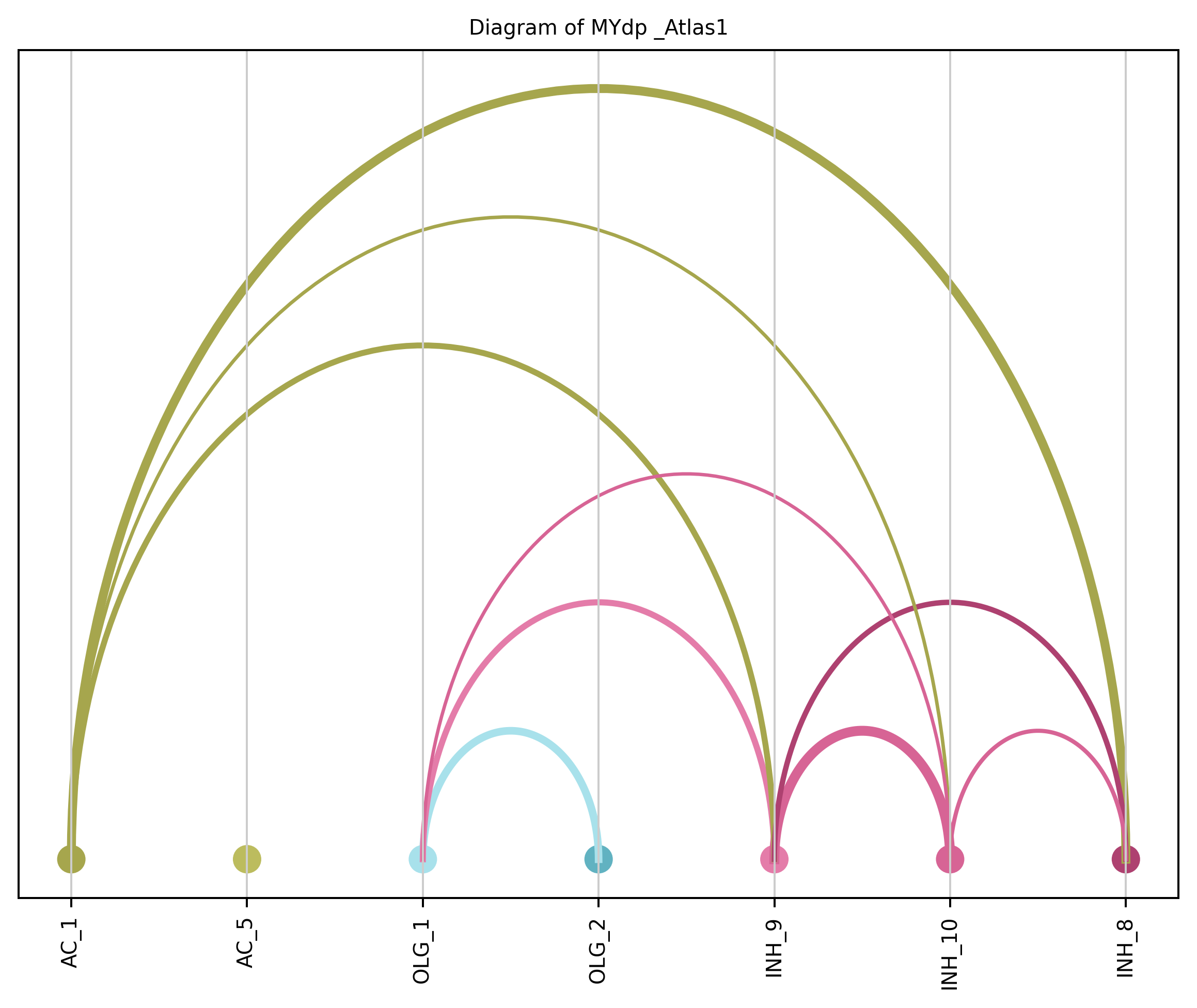
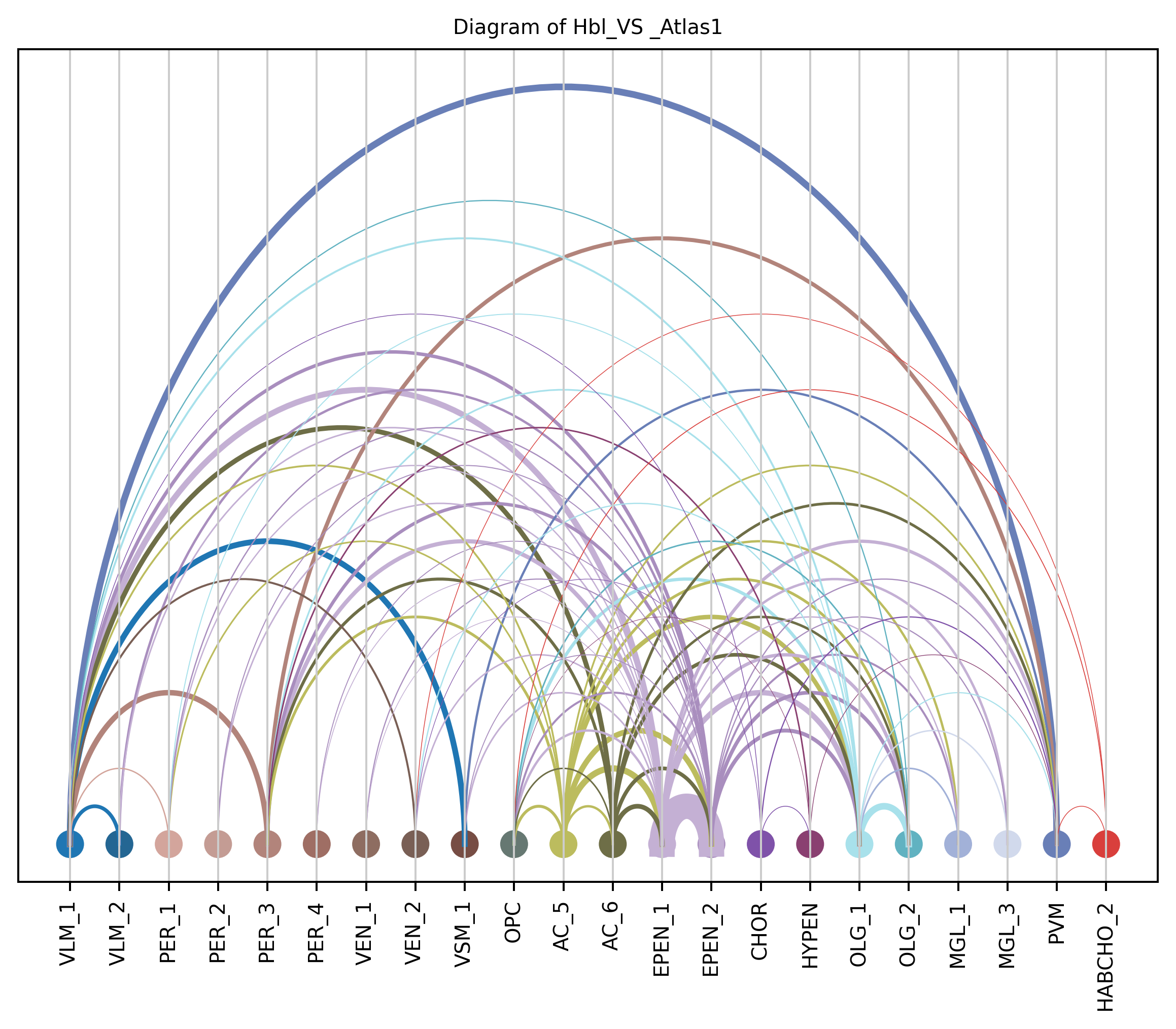
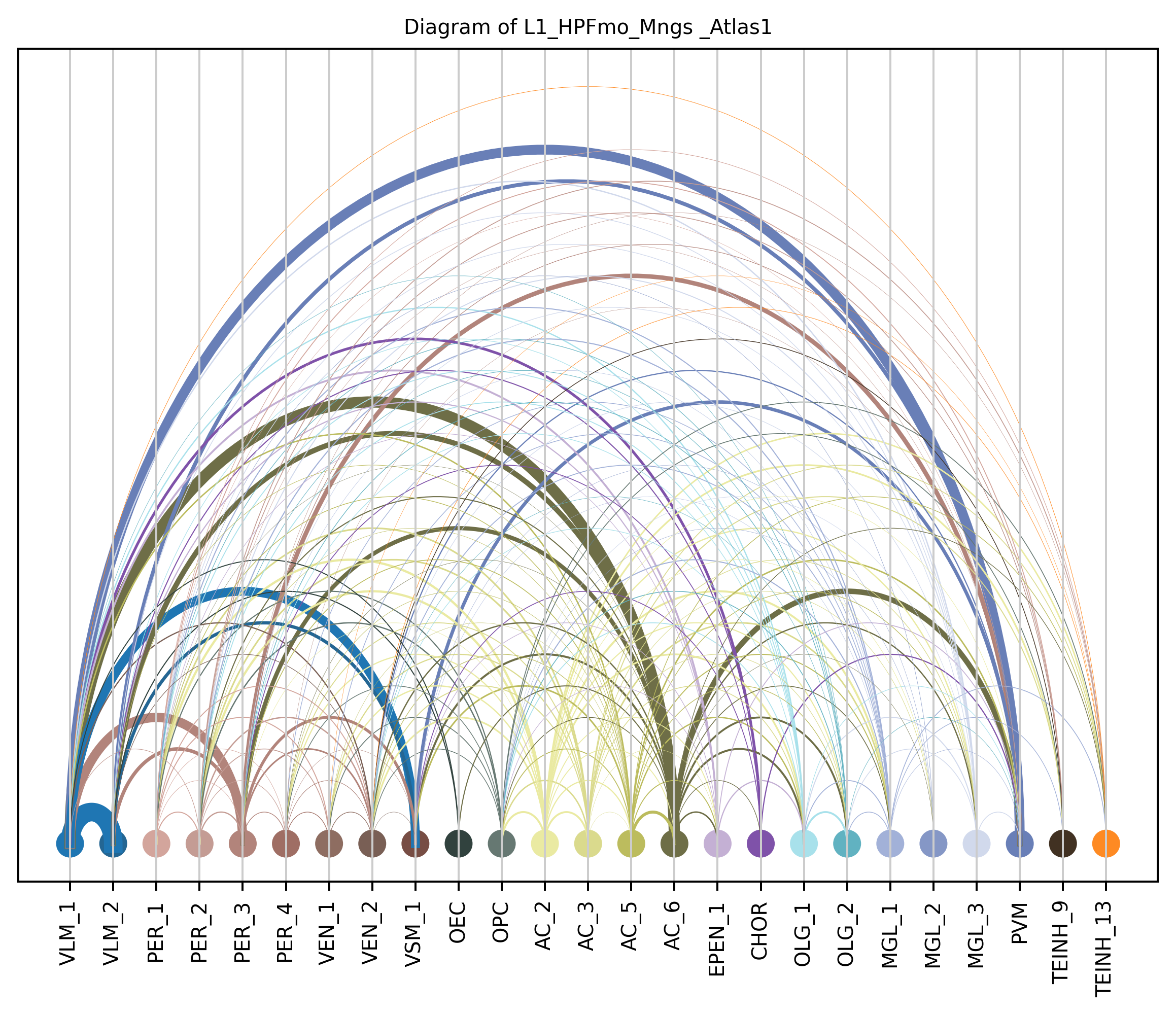
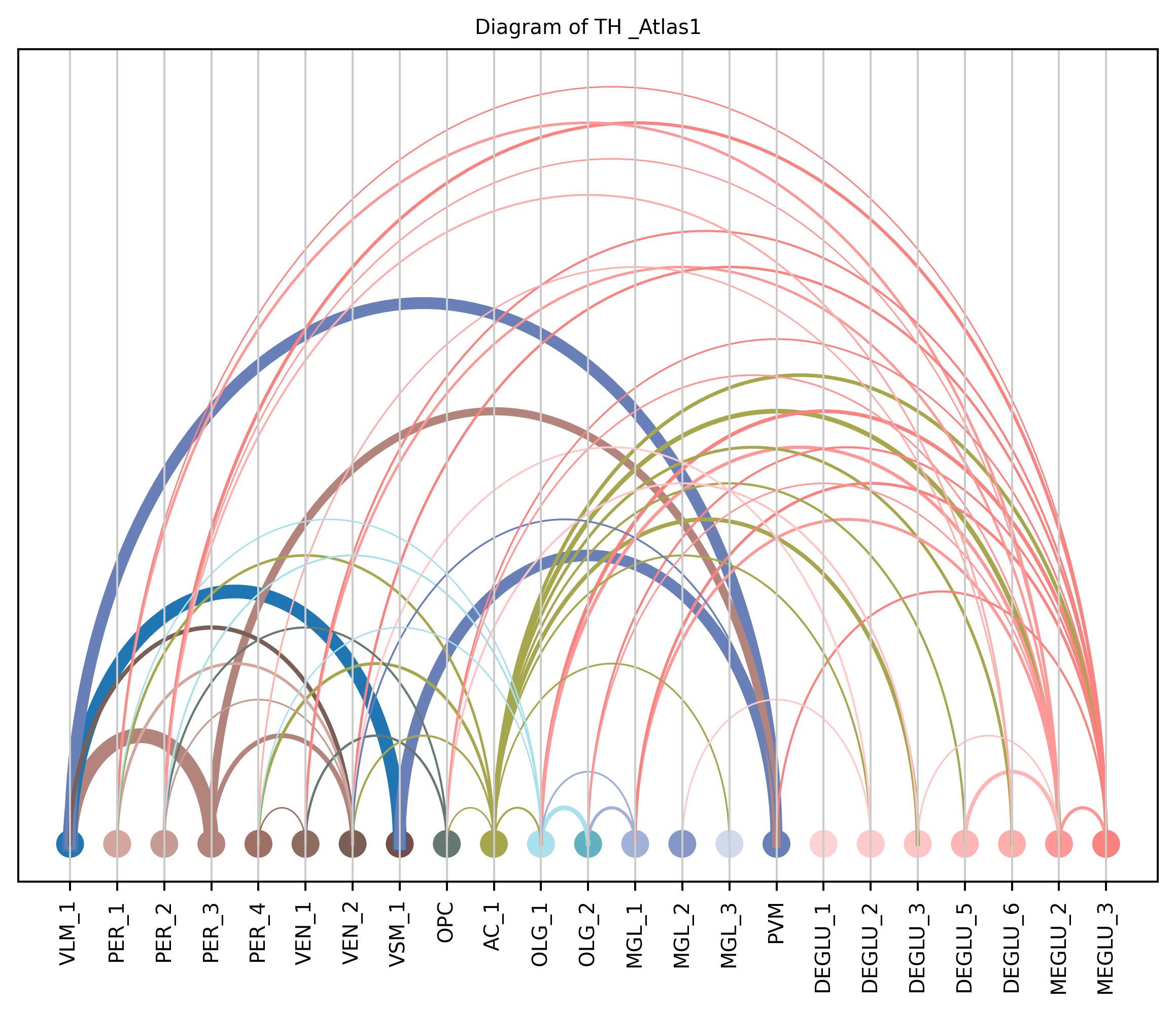
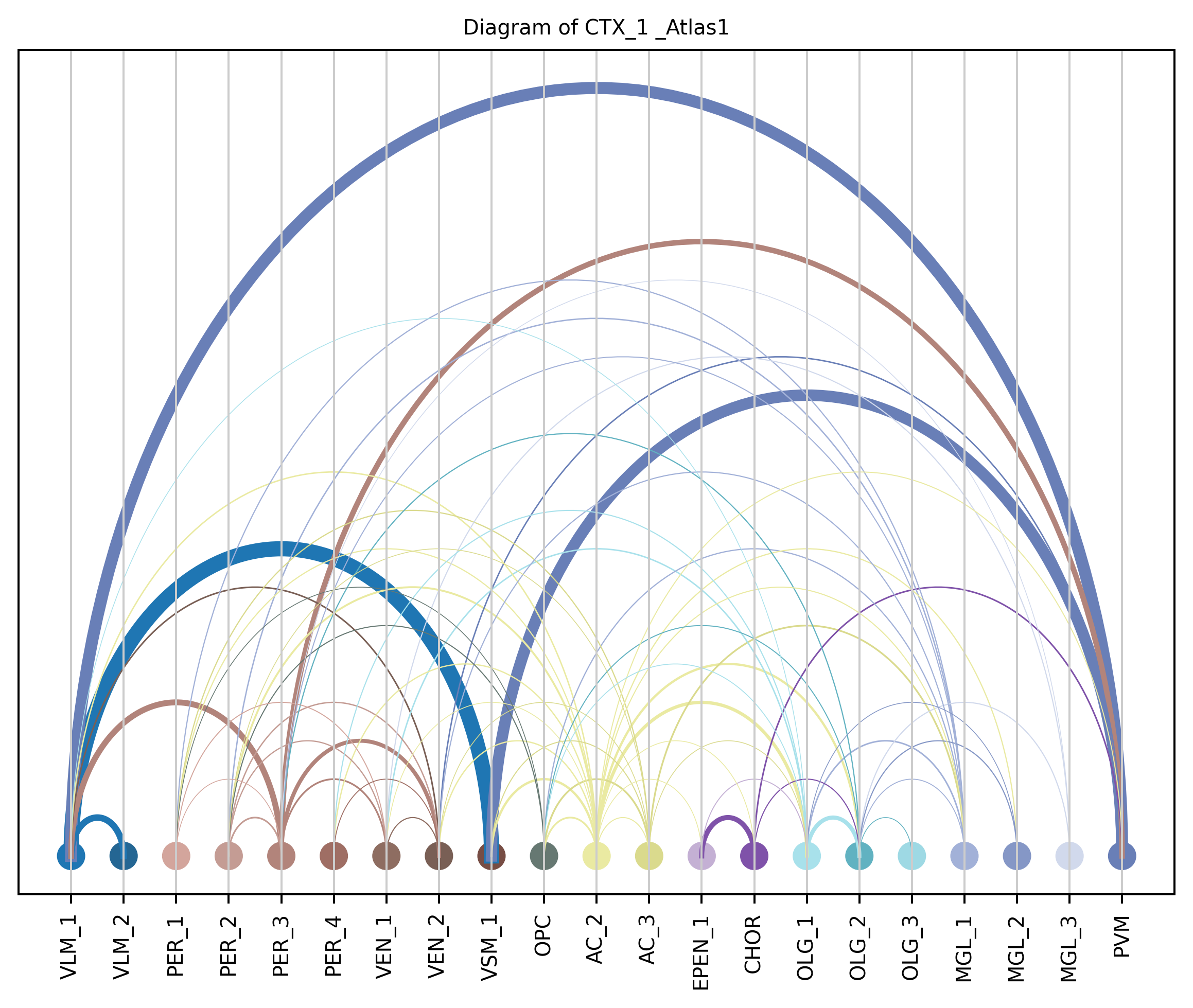
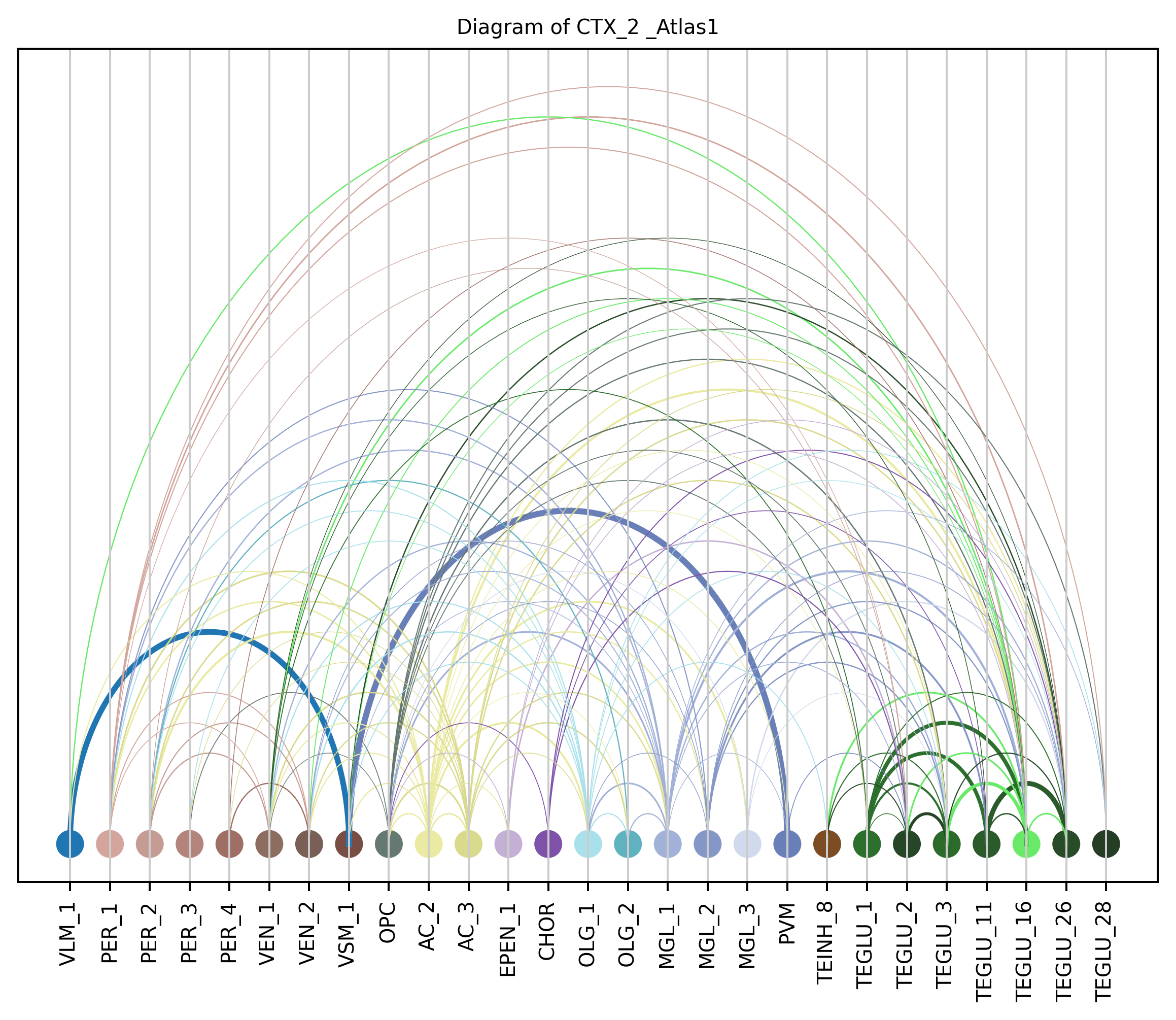
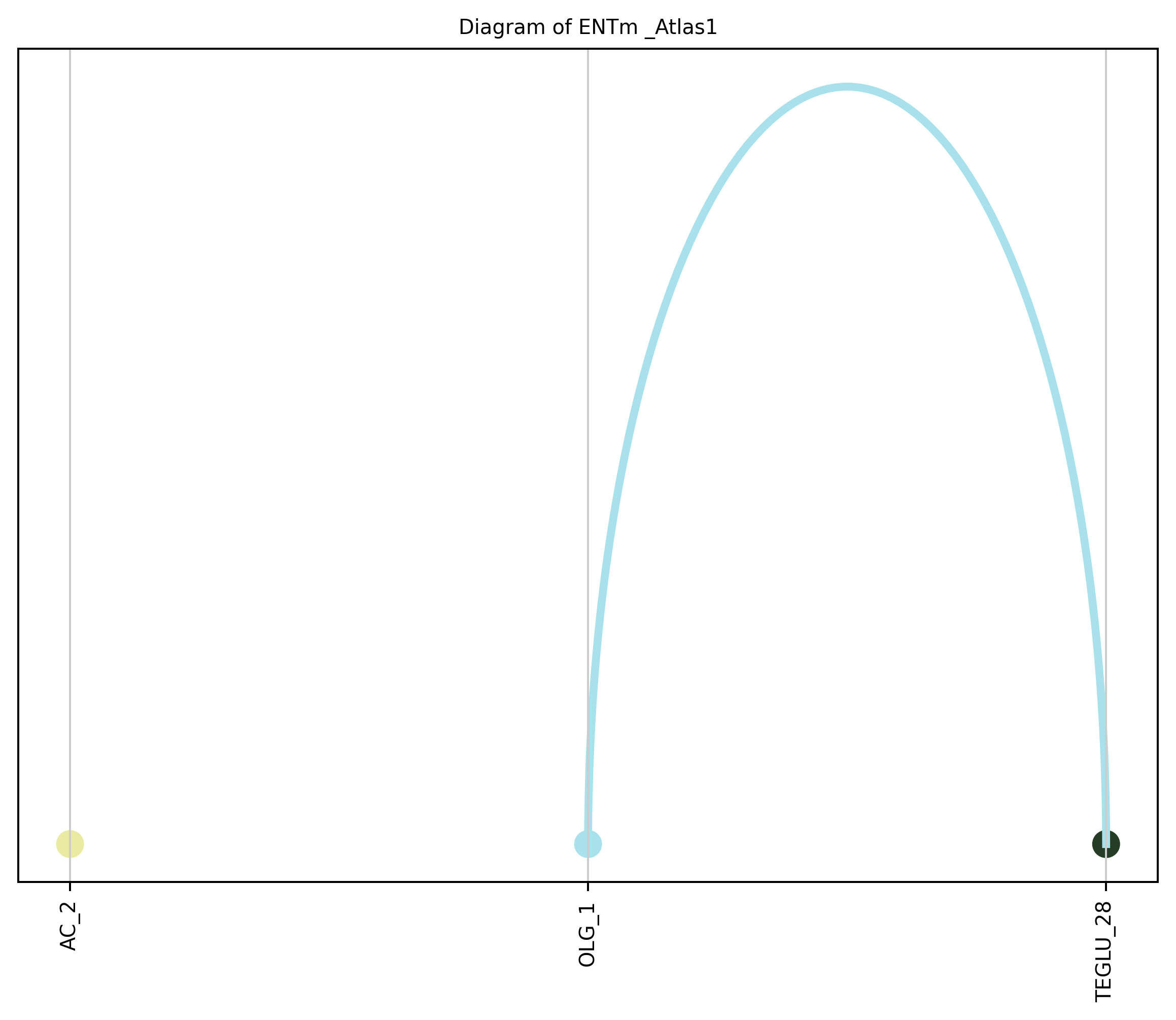
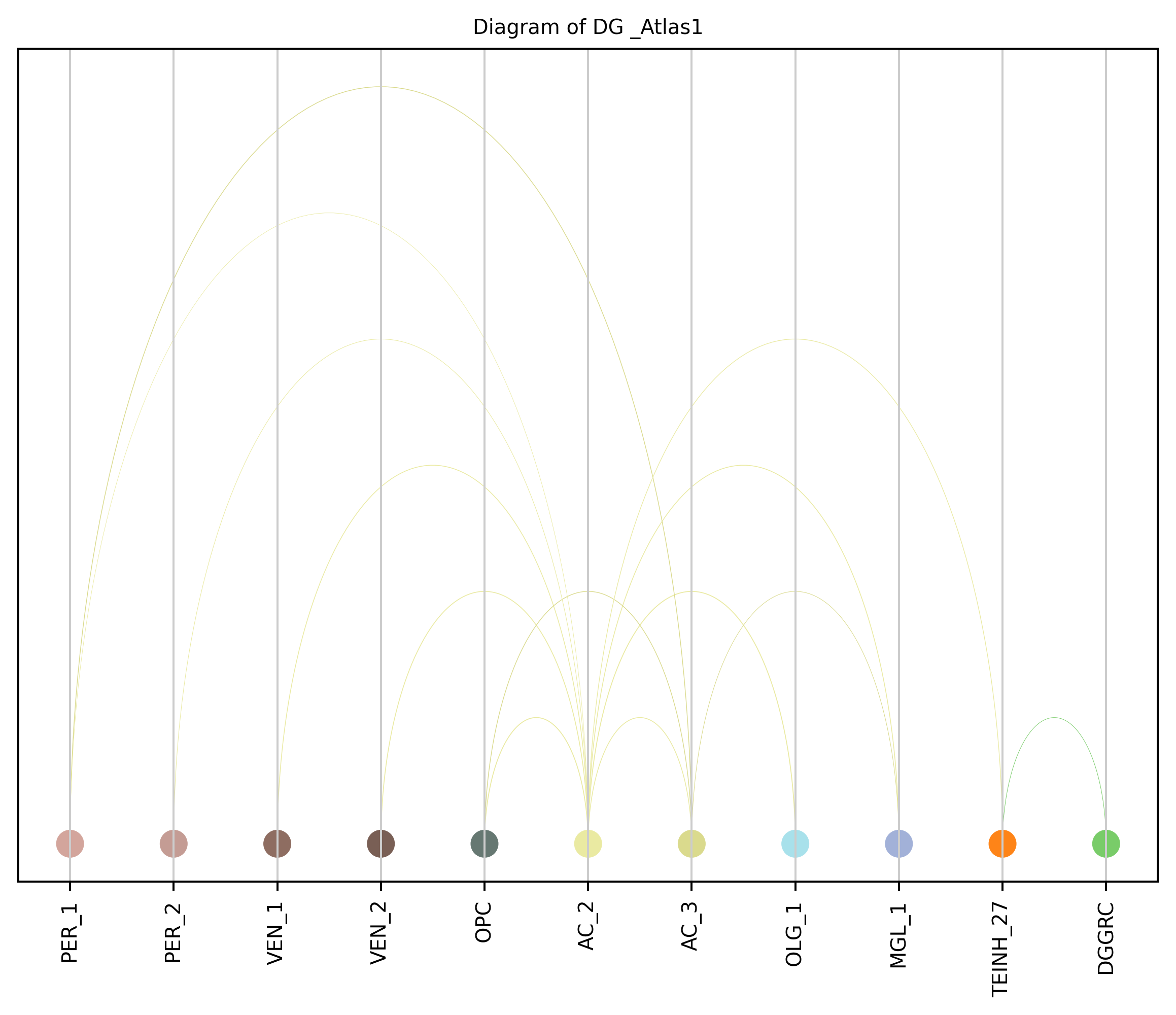
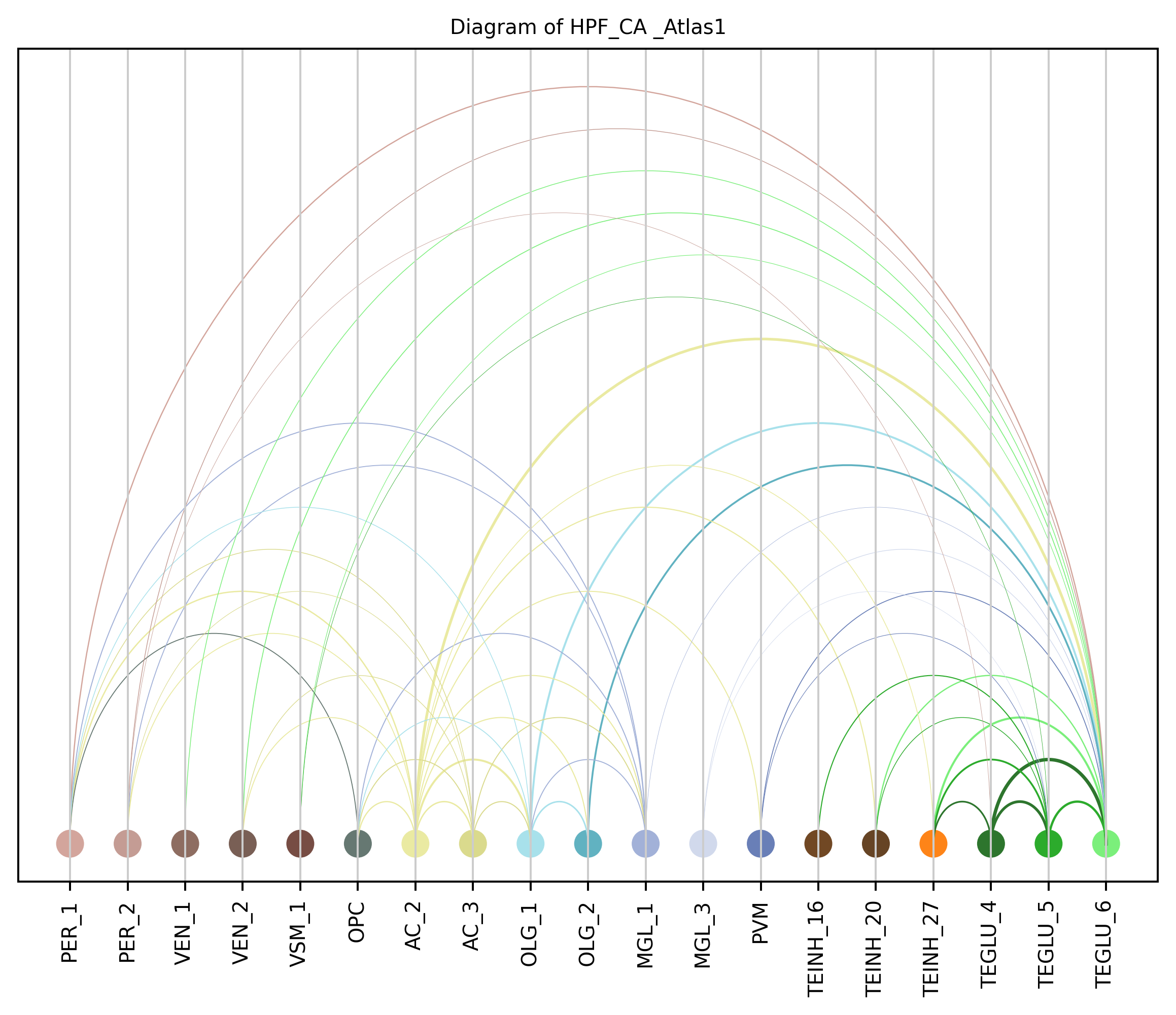
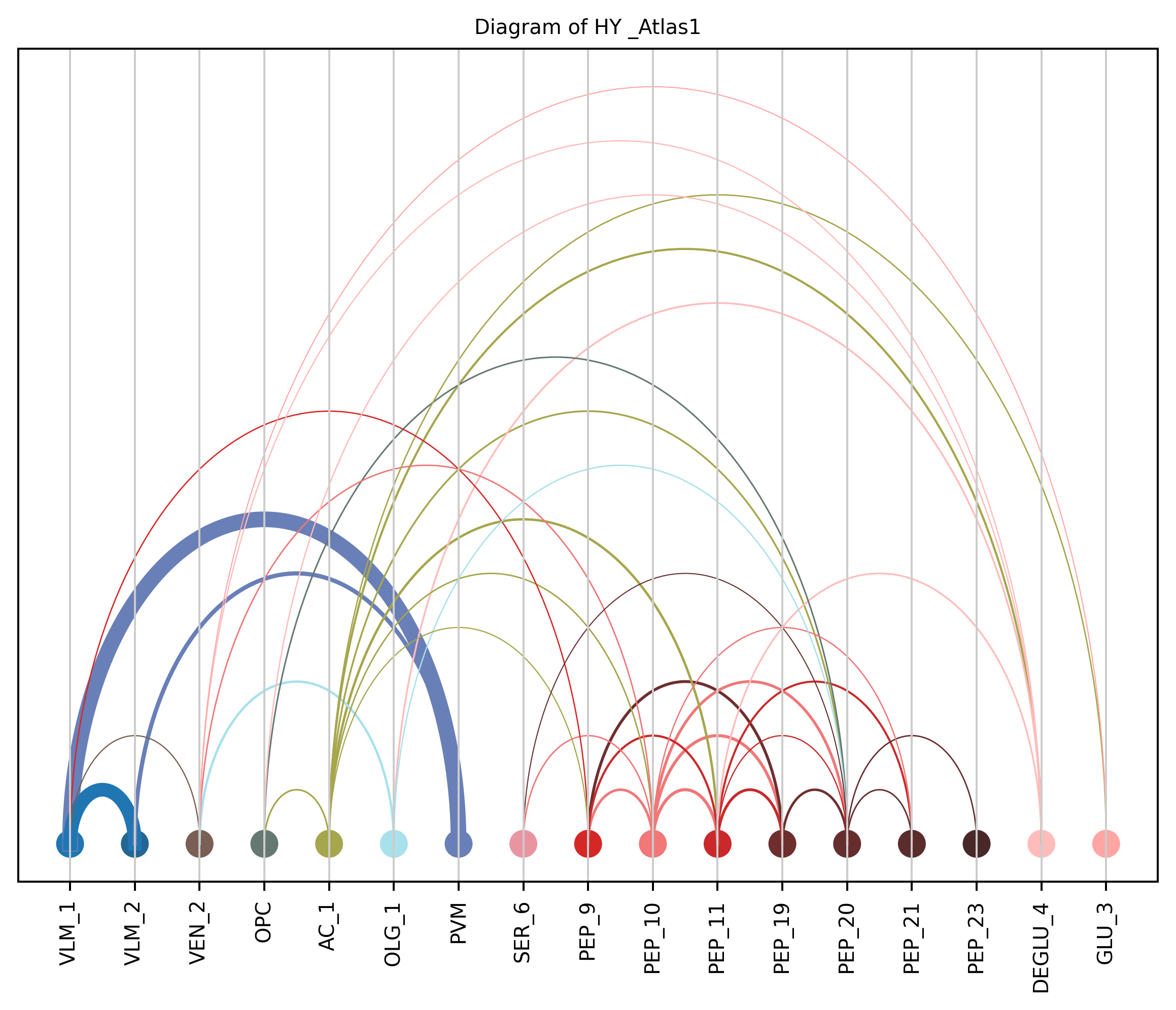
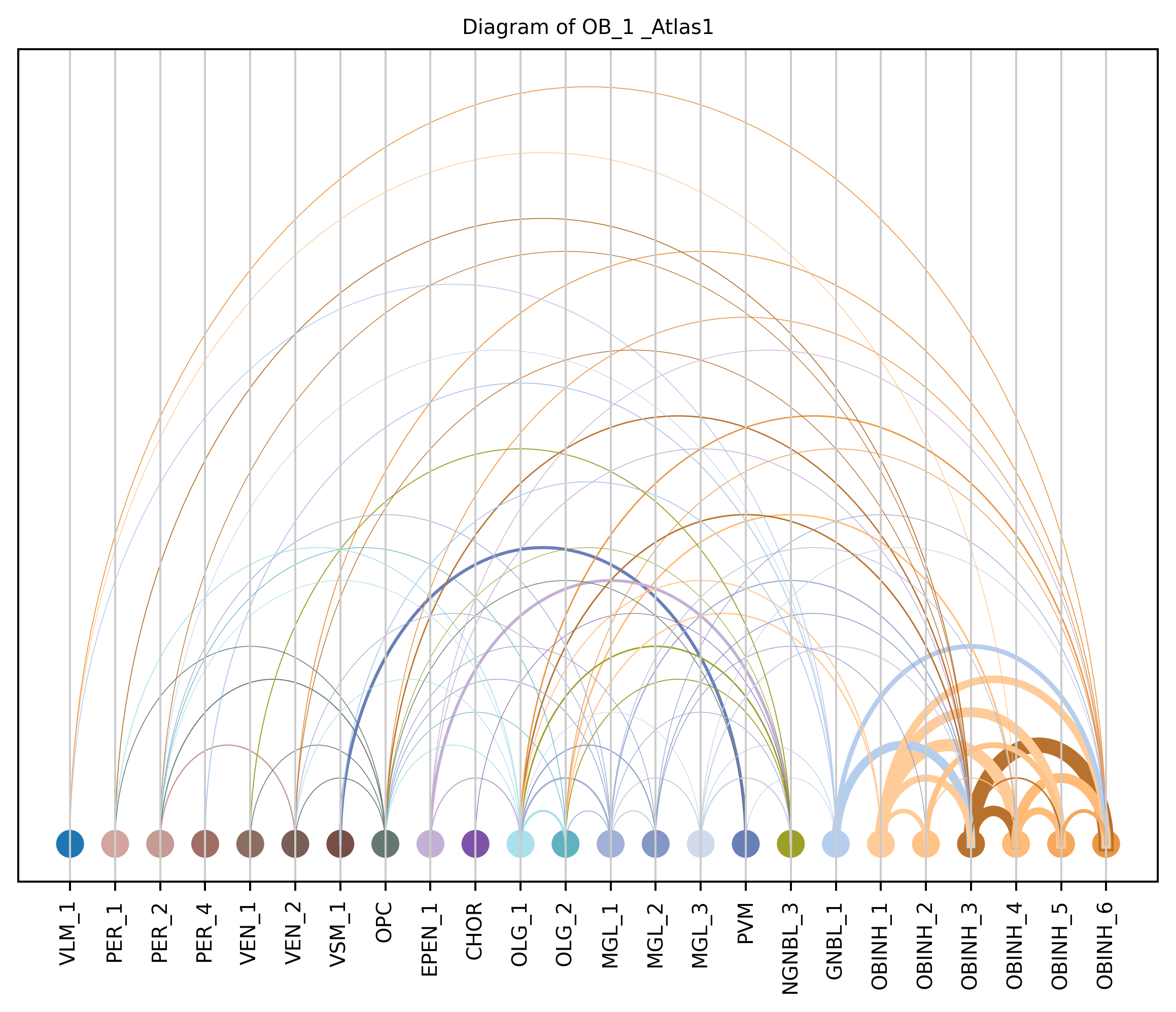
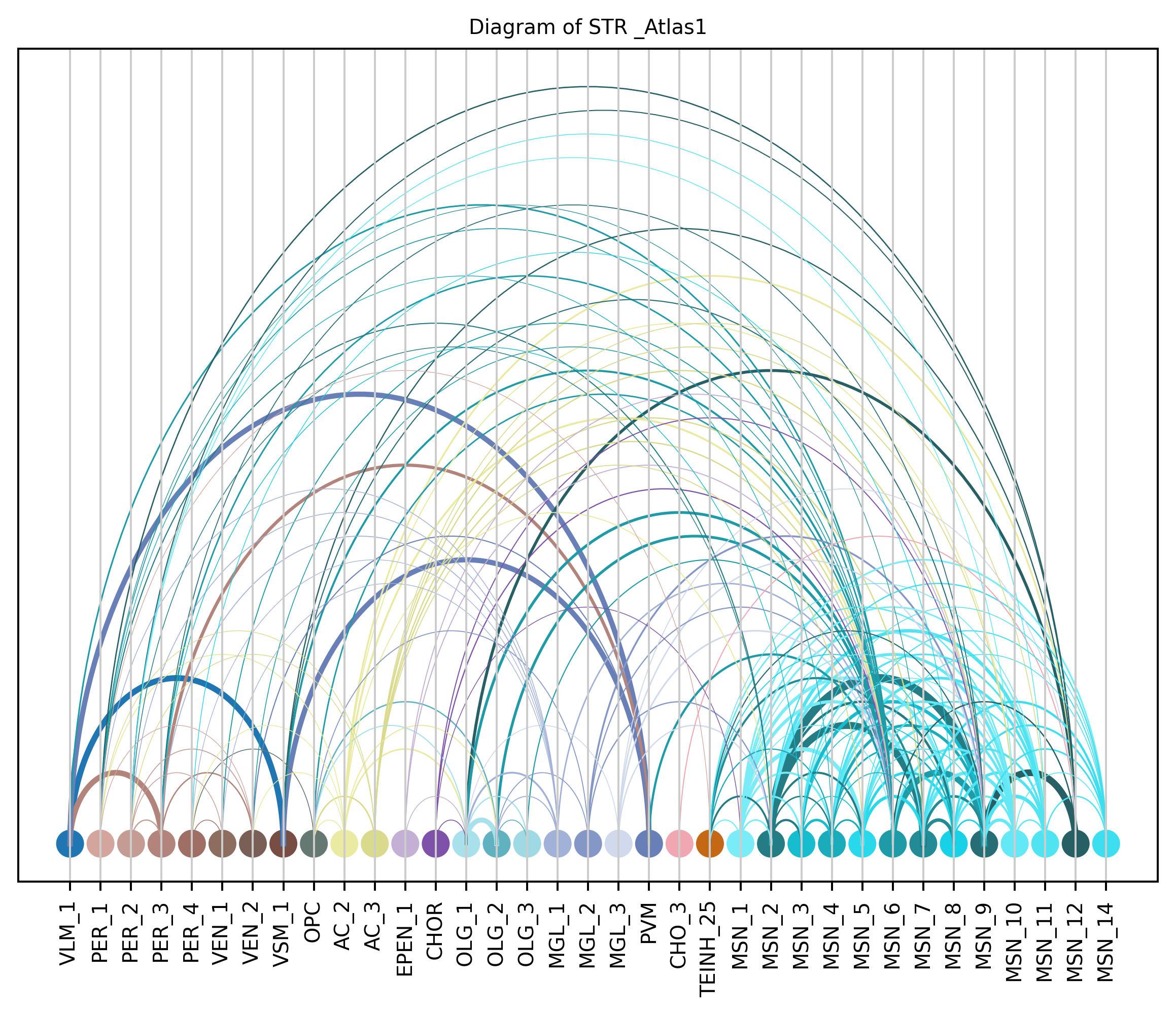
4. Plot arc diagram#
[13]:
import sys
sys.path.insert(0, f'{SOURCE_DATA_BASE}/arcplot-main/')
from arcplot import ArcDiagram
def NormalizeData(data):
return (data - np.min(data)) / (np.max(data) - np.min(data))
[15]:
color_tissue=pd.read_csv(f'{SOURCE_DATA_BASE}/color/starmap_sub.csv')
dic_color={}
for key,color in zip(color_tissue['key'],color_tissue['color']):
dic_color[key]=color
[19]:
for atlas_source in ['_Atlas1']:
permutation_path_merfish=f'{SOURCE_DATA_BASE}/outputs_30um{atlas_source}'
combined_results=pd.read_csv(os.path.join(permutation_path_merfish+'/result/', 'all_close_contacts.csv'))
save_path=os.path.join(permutation_path_merfish+'/figures_pvalue/')
os.makedirs(save_path, exist_ok=True)
for region_i in major_brain_regions:
combined_results_CB=combined_results.loc[combined_results['region']==region_i]
if combined_results_CB.shape[0]<2:
continue
# print(region_i)
combined_results_CB['from'] = combined_results_CB['cell_type1']
combined_results_CB['to'] = combined_results_CB['cell_type2']
pvalue=combined_results_CB['z_score']
combined_results_CB['weights'] = pvalue
transparency=np.array(-np.log10(combined_results_CB['pval-adjusted']))+10
transparency[transparency==np.inf]=350
transparency = NormalizeData(transparency)
transparency[transparency<0.7]=0.7
combined_results_CB['alpha'] = transparency
combined_results_CB_link =combined_results_CB[['from','to','weights','alpha',]].copy()
def createArcDiagram(df, node1, node2, weights=None, alpha=None,
bg_color='white',
cmap='viris', title=f'Diagram of {region_i} {atlas_source}'):
# get all the nodes
nodes_old = df[node1].unique().tolist() + df[node2].unique().tolist()
nodes_old = list(set(nodes_old))
nodes=[]
for i in dic_color.keys():
if i in nodes_old:
nodes.append(i)
custom_colors=[dic_color[i] for i in nodes]
# create the diagram
arcdiag = ArcDiagram(nodes, title)
arcdiag.set_custom_colors(custom_colors)
arcdiag.set_label_rotation_degree(90)
if not weights:
df['weights'] = 0.1
# connect the nodes
for connection in df.iterrows():
arcdiag.connect(
connection[1][node1],
connection[1][node2],
# transparency=connection[1][alpha],
linewidth=connection[1][weights],
)
# custom colors
arcdiag.set_background_color(bg_color)
# plot the diagram
# arcdiag.show_plot()
arcdiag.save_plot_as(f'{save_path}/{region_i}.png', resolution=300) # for saving file as an image with an optional resolution setting for higher-quality images.
createArcDiagram(
combined_results_CB_link,
node1='from',
node2='to',
weights='weights',
alpha='alpha',
)
[ ]: